

First, do no harm - how the FDA crippled America's response to COVID-19

Originally published August 2021, I’m republishing this now as I am migrating old blog posts to my substack and it is still very much worth listing all the ways the FDA failed at COVID.
This is a partial account of how the FDA and the medical establishment failed America in fighting COVID-19, and how the inherent structure of the FDA made the pandemic worse. They prevented individuals and companies from creating and providing timely drugs and tests to combat COVID-19, contributing to the deaths of hundreds of thousands of Americans and trillions of dollars of lost economic output.
This was not a one-off failure of the FDA. Since the modern FDA was created in the 1960s it has exacerbated epidemics and slowed life saving medical progress. COVID-19 cast into sharp relief the systemic inadequacies and costs of the FDA, but even in 'normal' times the existence of the FDA causes a great deal of unnecessary death and suffering.
The impetus for this post was a conversation I had with a friend during that interminable, locked-down summer of 2020. In the midst of discussing the US's pandemic response, I expressed anger at the failure of the FDA, to which he expressed genuine incredulity. "These are some of the smartest people in the world, if they're failing it's because it's an impossible problem!" (paraphrasing).
After some mutual-but-maybe-more-me yelling, on reflection, I understood where he was coming from. The FDA staff has decades of experience in medical research, and a six billion dollar annual budget; why would I think that they were performing worse than anyone else in their position could perform?
My response at the time was to gesticulate wildly, waving my hands around to capture the full length and breadth of the state of the country. But I think the question deserves a longer answer.
The following essay is that answer. The FDA as it is structured today is not up to the task of managing our medical system - the centralized, single reporting structure they enforce slows down the entire US medical system, failures that have been on full display with COVID-19.
We should break up the functions of the FDA and create a more robust, decentralized process with competing government and non-government licensing bodies.
Setting the Stage
The FDA, or Food and Drug Administration, is a federal agency responsible for approving drugs and medical devices. They also monitor and regulate food safety, but we're only going to focus on the medical part of their remit.
The received wisdom on the FDA is that they're the best of the best. To quote from the FDA website:
Today, the drug review process in the United States is recognized worldwide as the gold standard.
The FDA as the pre-eminent medical regulatory board seems widely accepted as true. The FDA 2018 budget was 5.1 billion, compared to 31.5 billion yen for the PMDA. I chose the PMDA as a comparison because Japan has the third largest economy in the world, and stats on China's allocations were hard to find. That being said the EU allocated 337 million (in USD) for the EMA in 2018. This isn't exactly a fair comparison, because the European Medicines Agency doesn't replace individual country regulators; however, both stats give a sense of the size of the FDA as a singular organization relative to other medical regulatory bodies.
Its significantly larger than international peers - for instance the FDA's 2018 requested budget is 20x greater than its Japanese counterpart.
The biggest pharmaceutical companies in the world are headquartered in the US.
Many governments modeled their own drug approval practices on those of the FDA.
And relative to the companies it regulates, the FDA is one of the most powerful government agencies. As Daniel Carpenter quoted from one Pharma CEO in the introduction to his excellent book on regulation and the FDA:
The FDA is standing there with a machine gun against the pharmaceutical industry, so you better be their friend rather than their enemy. They are the boss. If you’re a pharmaceutical firm, they own you body and soul.
The FDA-as-Scarface imagery is because the FDA is the sole gatekeeper for the entire, $500 billion+ US pharmaceutical market. They certify which drugs can be sold, determine how medical trials need to be run, evaluate the purported effectiveness of a treatment, and in the end have the singular power to reject any drug or medical device from being sold or used in the US (and often by extension, the world).
There is little to no recourse if a drug is rejected; if they don't like it or you, you're done.
No other regulatory agency in the US has this level of power. No other government body has the singular power to determine the fate of multi-billion dollar products.
From this power, the FDA shapes the culture of medicine and medical science in the United States. Quoting again from Carpenter:
It is fair to say that the basic terms, standards, schedules, and rules of modern drug development have been fashioned by the [FDA] as much as by any other global entity. When scientists and physicians test a new drug in clinical studies separated by “Phase 1,” “Phase 2,” and “Phase 3”; when companies submit a study “protocol” to the Administration that defines hypotheses and measures before their assessment and use; when firms and physicians debate the “efficacy” and “safety” of a drug before its approval or afterward; when scientists attempt to demonstrate the “bioavailability” of a drug in a given dosage form; when legislators write laws and insurers write policies governing generic drugs that depend upon demonstrations of “bioequivalence”—in these cases and many, many others, human agents are consciously and unconsciously using terms that have been deeply and thoroughly shaped by officials of the Food and Drug Administration. In essence the FDA sets the rules for public health policy and process.
How did the FDA gain such a preeminent position? The FDA traces its history to the late 19th century. The Division of Chemistry, under the US Dept of Agriculture, investigated food and drugs and published advisory notices on adulterants in food. In the first half of the 20th century, as part of a progressive trend empowering government agencies, the FDA began to regulate and certify more of the pharmaceutical industry. Often this was in response to tragic events where improper drug manufacturing practices led to poisonings and death. By the 1950s the FDA was responsible for certifying drugs were safe before they could be used in the United States.
However, there was a 'discontinuity event' with the FDA's powers; while before they were but an actor in the pharmaceutical ecosystem, afterwards they would fully control it.
In the early 1960's a new drug, Thalomide, was developed and used in Europe to treat pregnant mothers' morning sickness. It had severe side-effects, leading to miscarriages and deformities in tens of thousands of pregnant women and newborns.
While Thalomide had not been approved for use in the United States - and in fact was still being assessed by FDA officials for safety - the public outcry in response to the horrifying reports coming from overseas demanded immediate action.
Prior to the side-effects of Thalomide being discovered, Senator Carey Kefauver of Tennessee had been holding hearings on the pharmaceutical industry. Sen. Kefauver was opposed to what he saw as the excessive profits of the pharma industry and their aggressive marketing, and he pushed for greater government control over industry practices.
At that time, the U.S. Food and Drug Administration had limited authority to require efficacy standards or disclose risks. Kefauver was accused of expanding the power of government excessively, interfering with the freedom of doctors and patients, and threatening the viability of the pharmaceutical industry. His legislation seemed likely to fail.
But, after Thalomide, Kefauver's amendment quickly gained political support. Even though it was not targeted at preventing Thalomide like events, and even though Thalomide had not been officially approved in the US, the urge to 'do something' was compelling, and the amendment sailed through.note: Carpenter disagrees with the view that the amendment represents a radical break in FDA power; his book has a wealth of details on the 'continuous' growth in the FDA's regulatory power in the 20th century. I think the magnitude of the change represents a break, but whether it's a continuation or discontinuity is not, in my opinion, a crucial distinction.
It's wrong to view the question of whether a drug is effective as having a single, stand alone yes-or-no answer. The question of effectiveness is deeply contextual, and different people and experts will have different takes on the value of a drug or medical device depending on the circumstances in which its used. The requirement to determine effectiveness, and to act as a gatekeeper for effective vs. ineffective drugs, dramatically expanded the discretionary power of the FDA. It also expanded the cost and size of clinical trials and reduced the total number of drug approvals. The graph below, from the FDA Review, details how the number of new drugs approved fell dramatically after the expansion of FDA powers with the 1962 amendment.
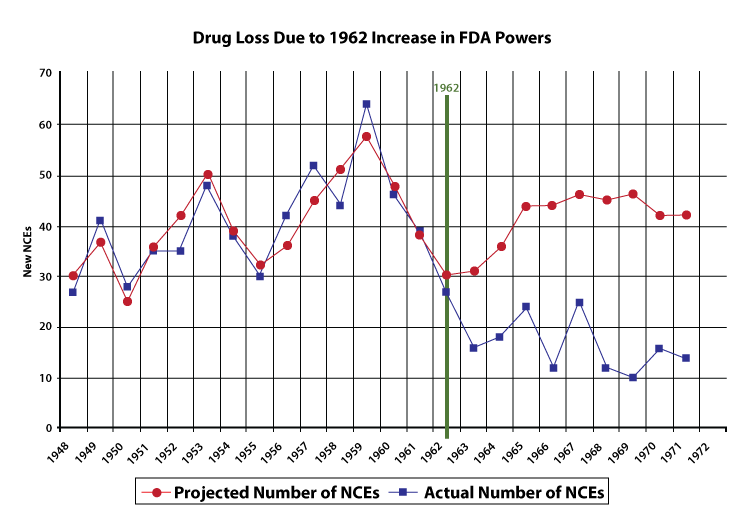
Supporters retort that empowering the FDA has prevented companies from taking advantage of people with ineffective drugs, that slower approvals mean safer approvals, and that the FDA, in its role as referee, has acted as a champion of good science.
In the last year these arguments, and the FDA's reputation, would be put to the test.
Initial Response
In late 2019, reports started to emerge of a potentially dangerous virus in Wuhan, China. While local Chinese officials tried to suppress the news, information leaked out as brave individuals warned of the threat.
In January 2020 the CDC created a test for what was now known as COVID-19, and submitted it to the FDA, who approved it. However, troubling reports started coming in of bad results and bad data - state labs had discovered that the CDC's test for COVID-19 had serious performance issues, compromising its diagnostic value.
By Feb. 8, hours after her lab started verifying the test kits, Rakeman said she began receiving panicked emails from colleagues. Something, they said, was wrong: They had run the tests dozens of times but were getting inconclusive results from two of the reagents, or chemicals, in the vials. Rakeman began calling other labs to see if they were experiencing the same problem, and they said they were. "It was very truly an 'oh, crap' moment," Rakeman said. "These reagents aren't working; everybody is waiting for us all over the city to have this test online. We think we have more cases than we've been able to detect and the test isn't working."
It was becoming obvious to everyone paying attention that COVID-19 was a problem, but it was unclear where it was, how quickly it would spread, and who had it - all of the pertinent details necessary for organizing a response. The FDA's approval of a flawed test - the only test that it had approved for COVID-19! - kept the country in the dark.
But, to err is human. To forgive and quickly get another test approved, divine. To instead prevent everyone else from getting anything done – well, now you're acting like the FDA.
Faced with the failure of the CDC's test, hundreds of universities, commercial labs, and individual researchers sprang into action and began working on creating their own tests and ways to scale up testing. It was the civic sector stepping up to help combat a country wide crisis.
But, instead of being greeted as heroes trying to fill the testing gap left by the failure of the CDC, they were greeted by a blizzard of paperwork and an icy bureaucracy that made it illegal to test for COVID-19.
This is because the FDA involves itself in all aspects of medical device development, and decisions like repurposing a flu test to check for COVID, or providing those results to patients, needed to be run past FDA officials.
So groups like the Seattle Flu Study, a group staffed by world-class researchers and physicians, with funding from the Bill & Melinda Gates foundation, had to hunt and beg for someone to grant them an approval to move forward, to use their functioning tests and labs to learn whether people were unknowingly spreading a deadly virus. They were repeatedly denied, because their lab was not certified as a clinical laboratory under existing CMMS regulations.
Let's pause for a second and ask the obvious question - should they have been allowed to test? On the one hand, we can look back and see that yes god yes wtf how is this even a question. Better tests would have helped mobilize resources and limited the spread of the virus in the early days of the pandemic.
But on the other, when judging policy, we're evaluating a decision rule - the general process that we follow when making a decision. In this case its the rule 'who gets to participate in the medical process and in what way', or more specifically 'who gets to test and for what purpose'. As we saw from the CDC and FDA's initial failure to approve a working COVID-19 test, faulty tests can have serious consequences, and following established procedure should (probably) reduce that likelihood.I have not found cost-benefit analysis on the procedures to approve medical tests and whether they do more to reduce the likelihood of faulty tests relative to other evaluation techniques
Given this, it becomes a question of 'what's the meta decision rule' - how do we know to throw out our existing process and do something different because the circumstances have changed. The ability to deviate from the script is the mark of a live player, aka someone or some group who can do something novel when its demanded of them. Reasonable people can disagree about when this call can be made, but it's a call that at the very least needs to be actually evaluated by someone.
The actions of the FDA revealed they couldn't do that, that in the end all they had were their existing scripts. Even in the wake of their failed test and increasingly dire reports of a plague spreading unchecked, they were unwilling to deviate from their regularly scheduled processes:
The Association of Public Health Laboratories, which represents American laboratories, made an unprecedented request to the commissioner of the FDA to make an exemption so that state and local public health labs could use the tests they had created. [the FDA commissioner] responded, stating “false diagnostic test results can lead to significant adverse public health consequences” and that the laboratories were welcome to submit their own tests for emergency authorization."
The emergency authorizations would take months.
Alex Greninger and his colleagues at the University of Washington “spent almost a hundred hours filling out a baroque, thirty-page form, filing the authorization request on February 19th. Still no dice: he had e-mailed the material, and the F.D.A. insisted that he print it out and mail a hard copy, along with the digital file in physical form, such as a thumb drive or a CD, to a separate ‘documentation’ office.” (New Yorker)
The FDA insisted on continuing with their existing lengthy bureaucratic processes and were intent on enforcing them, regardless of the real world costs. This graph, looking at the rate of testing in January and February, really says it all, demonstrating the artificial cap on testing imposed by the FDA when outside labs were prevented from participating until the policy was loosened in March (far too late).

source: Center for Growth and Opportunity
Contrast the behavior of FDA officials with the South Korea government.
On Jan. 27, the South Korean health officials convened experts and test-kit companies at a conference room inside Seoul Station, an easy central location for experts strewn across the country. They got an early surprise. Two South Korean firms had already begun developing their own tests.
“We promise a fast approval,” said one South Korean health official at the time, saying the government would buy unused supply if the outbreak never reached a significant size. One firm, Kogene Biotech Co., demonstrated a successful test and got the regulatory green light within four days.

They mobilized a country wide testing regime in weeks, an order of magnitude larger than the US.
In the US, on the other hand, creative solutions from the public and private sector to expand access to testing were either never implemented or put into action too late to be useful. Consider pooled testing, an approach for maximizing detection of the virus in a population while conserving the number of tests used. It could have 10xed the throughput of the US testing regime. But this creative tactical approach was delayed until the point of uselessness - emergency authorization for pooled testing from the FDA was only given in mid July. By that time, as the NYTimes reports, it was too late:
But now, when the nation desperately needs more coronavirus tests to get a handle on the virus’s spread, this efficient approach has become worthless in many places, in part because there are simply too many cases to catch.
The FDA didn't act at the speed the country needed them to, and seemingly were unable or unwilling to adjust their procedures to account for the crisis. They were a dead player.
Bottlenecks
The failure to test would continue throughout 2020. America drastically underproduced the number of tests that the situation called for. The mismatch in demand and supply meant that the results of a test could take days or weeks to be returned, by which point an infectious person might have already spread the virus to others.
And while there were many proposals to 'flood the zone' with tests, we never got to the point where they were truly widespread, in large part because the FDA continued to follow and enforce their existing controls, which constrained the supply side of testing.
For the default, PCR COVID tests, it was a multi-stage process with many potential bottlenecks:
You needed a nurse or doctor to swab your nose and collect the sample. If they were all occupied swabbing other peoples noses or, you know, treating the sick, you couldn't get your test run.
There were a limited number of processing centers where the tests could be run, and they quickly got swamped by demand.
Reagents were in short supply for the reactions. So were PCR machines. If either of these weren't available, then the tests wouldn't be run.
These are all artificial bottlenecks. For instance, allowing regular people to swab their own or others' noses (I can assure you it doesn't take four years of medical school to do this), or allowing new manufacturers to enter the reagent market, could have dramatically expanded the testing regime.
There's no more striking example of this artificial constraint than the failure of the US to leverage rapid testing.
Rapid tests provide quick, immediate feedback on whether someone is contagious. If a person has COVID-19 and can spread it to someone else, a rapid test will tell you that in minutes. While they are less accurate than PCR tests, they are more effective at the public health goal of detecting if someone can spread the virus.
And that is, self-evidently, the most important information a person needs when determining, for instance, whether to go to work or stay at home and self-isolate. Not only are rapid tests faster, they're much cheaper, cheap enough to be mass produced for everyone in the country.

This was the type of testing technology that could have dramatically changed the course of the pandemic. As Michael Mina, an advocate for rapid testing wrote:
The U.S. government can produce and pay for a full nation-wide rapid antigen testing program at a minute fraction (0.05% – 0.2%) of the cost that this virus is wreaking on our economy…These tests are incredibly sensitive in catching nearly all who are currently transmitting virus. Widespread and frequent rapid antigen testing (public health screening to suppress outbreaks) is the best possible tool we have at our disposal today—and we are not using it.
And we didn't use this tool, despite it being invented in April, because the FDA had a single pathway for approving testing, and it didn't account for the value of speed or cost. Instead they insisted on treating them like PCR tests. Because PCR tests are more accurate and have a higher likelihood of detecting viral load, on this single dimension PCR tests will beat rapid tests.
But that doesn't matter if you're trying to detect who is spreading the virus! Forcing rapid tests to follow the same 'assessment' path as PCR tests meant that approval was delayed and delayed as the FDA treated this public health tool like a medical device and forced it to meet similar standards; it was only in December that they were approved, and in a crippled form that required a doctor's supervision, decreasing their usefulness as a population level tool.
Imagine this. It’s September 2020. Rapid tests are available through a subscription service sent right to your door every day. You feel fine but you decide to go over to a friend’s house and take a test first, and ask them to take one too, knowing with confidence that given the negative result you’re almost certainly safe to hang out. And should you start to feel sick, you can find out quickly and avoid others getting sick.
This isn’t some imaginary sci-fi world, this is the kind of technology that could have been available right then! All it would have needed was imagination on the part of regulators.
Vaccines
The fastest vaccine development ever carried out has turned the tide on the pandemic and saved millions. It's a triumph of modern science.
But, they were only a triumph in that external actors overrode the default instincts and approach of the FDA. In many ways, the real triumph was cutting through the bureaucracy that normally accompanies new medical technology; it should serve as a massive rebuke to an organization that - even after the initial vaccine had already been developed - promoted an 18+ month timeframe for rollout.
The FDA repeatedly added stumbling blocks to the vaccine development and deployment, trying to turn operation warp speed into operation '30mph is good enough for government work'. Their day to day operations betray a culture that couldn't truly get behind moving at the speed necessary to save the most lives.
A few examples
Clinical Delays
The AstraZeneca vaccine candidate was paused twice, once in July and once in September due to patients falling sick from a neurological illness, Transverse Myletitis.
And of course people getting sick is important to investigate - that's the point of clinical trials - but the actual incidence was two out of 18,000 people. Of those, one already had a chronic illness that caused an increase risk of TM. So, to investigate a 0.0001% incident rate, they paused a clinical trial for a vaccine that was necessary combat a global pandemic that was killing thousands a day. In total it was months of additional time for the AstraZeneca vaccine, a far longer delay than other regulators took when investigating the trial, and a costly delay for the potential information gain.
With 18,000 data points, a “1%” risk would require there to be 178 missed cases. Odds of that are astronomically low. Much more likely, if there were any real correlation (which again seems rather unlikely) we are looking at something 80+) and would STILL result in fewer deaths than waiting.
For other vaccines, the FDA altered the analysis requirements, adding more time to the approval process. With Pfizer's vaccine, they asserted that they would need more data before a public analysis of the data could be conducted:
The FDA career staff also delayed the vaccine by adding an unprecedented requirement to slow down Phase III trials… the move was a departure from a decades-old standard operating procedure at the agency and the process used to authorize convalescent plasma just months ago.
The most shocking delay was, in the home stretch, when Pfizer submitted their clinical data for final review and approval, it took the FDA weeks to schedule and hold the approval meeting.
Pfizer submitted data detailing the safety and effectiveness of its vaccine on Nov. 22. But rather than immediately convening experts, the FDA scheduled a review meeting on Dec. 10, almost three weeks later. As Pfizer’s application sits on the shelf at the FDA awaiting authorization, about 27,000 Americans will have died. So what is the FDA doing for three weeks?
This pushed the vaccine release date back another month. During the period when the vaccine could have been approved, COVID deaths were the leading cause of death in the country.
First Doses First
After the vaccines were approved, there was a crucial choice facing countries - who should receive them first? While there were important choices around which segment of the population to receive them, a hidden assumption went unchecked for far too long - for vaccines that 'require' two doses, should we instead give out first doses to as many people as possible before the second dose?
The answer to that question could have significant ramifications, as it would determine the rate at which the population approached herd immunity.
However, it was a question that the FDA was, at first, uninterested in. Because the clinical trials had been carried out with two doses, that was, as far as the FDA was concerned, the only proper way to administer them. To change the administration protocol was beyond the pale.
Only after massive public campaigning was this re-evaluated in the US, and the trend continues, with the FDA and other regulatory bodies ignoring the potential benefits of fractional dosing.
Johnson and Johnson
While vaccine hesitancy was often referenced as the reason behind the 'safety first' attitude of the FDA, the J&J saga is illustrative. Unlike the Pfizer and Moderna vaccines, which used the novel, mRNA technique, up until March 2021 if you were - rightly or not - concerned about this approach, you could use the more traditional J&J vaccine.
I said up until March 2021, because at that point the FDA decided to be extra cautious and destroy the reputation of this vaccine.
However, after a series of reported blood clots, the FDA paused administration of J&J. Eight million people got the J&J vaccine - 28 cases of blood clots in total were identified in people who received that vaccine, with three deaths. There was no clear causal link, but the FDA played it safe. What did this additional safety buy in risk reduction?
I like the micromort method of evaluating risk - every micromort is 1 in a million chance of dying. So when you do an activity that is risky - pro tip, that’s everything - you incur a certain number of micromorts.
Activity Micromorts Skydiving 10 Swimming 12 Living for two days in New York City 2
Even if the J&J vaccine was the trigger, thats a micromort of 0.375. You’d need to inject yourself with 32 J&J shots to approach the risk of a day at the pool.
And just existing in America in 2020 exposed you to 0.5 micromorts from the risk of COVID-19! Orders of magnitudes more people would have needed to be getting sick to justify this delay - and the delay ended up destroying public confidence in the vaccines, which shows up in the sudden drop in vaccination rate.
It's crucial to keep in mind that during all of these delays and concerns about safety, we were actually administering another drug called 'lockdown'. For an organization that was monomaniacally focused on proper safety testing, this other intervention wasn't tested by the FDA and the side effects weren't evaluated. Those side effects included:
Trillions of dollars of lost economic activity
Destruction of human capital and social capital
A huge amount of general suffering
A tragic number of deaths
This was a particularly bad course of treatment, given potential alternatives.
Human Challenge Trials
The lengthiest part of the vaccine development process was clinical testing.
What if we could've skipped it entirely?
What if we could've engaged in a highly risky process, in which 10,000 volunteers risked their lives to save millions of people, perhaps in exchange for a handsome paycheck and hero status. Imagine in this scenario that their chance of death is 10%.
It seems like this would be the obviously moral thing to do. Not forcing anyone to take the vaccine, but offering civilians the chance to save the world, knowing all the risks involved.
Oh, and it wouldn't have taken ten thousand people. Maybe a thousand - with a chance of mortality of ~2%.
And embarking on this process – human challenge trials – we could have had the tools to beat the pandemic in Summer 2020. We simply lacked the will, or whatever it is that motivates some people not to wait weeks to schedule a meeting to approve a life saving drugs.
After a few weeks to months, needing only a few hundred volunteers, the world would have had strong evidence on whether a vaccine worked. Different dosage regimes and different viral exposure levels could be tested, which could have optimized the vaccine protocols (we'd actually know with confidence whether one dose was enough!). Contrast this with the approach that was used, where tens of thousands needed to participate over months, long enough that researchers can be confident that they can detect statistical differences in the infection rates of the two groups.
If we had run human challenge trials, we could have known in late spring or early summer of 2020 that the vaccines worked as well as they did. This would have:
Enabled an earlier rollout of vaccines.
Generated invaluable information about the virus and vaccine efficacy.
Rallied the public to take greater short term precautions with the confidence that vaccines would be coming.
The list goes on.
And the public at large supported the idea - in a survey 75% of people, from broadly different backgrounds, support human challenge trials. And it's worth noting that the leading consequentialist and deontological ethical philosophers, along with many others, signed an open letter calling for human challenge trials.
However the FDA declared that they wouldn't accept human challenge trials as evidence for vaccine development, and as discussed there's no alternative to the FDA, so that was that. The FDA's singular veto power meant there wasn't a path for challenge trials.
Monopolies
But can we conclude from this that the FDA was destined to fail? If the right person had been at the helm of the FDA, would everything have turned out differently?
It's a tough counterfactual to evaluate - I think its fair to say that a better leadership team, one attuned to the crisis and willing to fight to implement an effective bureaucracy, would have done on the margin better. But such a question presupposes that we ever could have had a truly effective FDA - their are systematic reasons why the FDA, even with the largest budget in its history and among the world, is as hollow as it is.
The structure and culture of the FDA is shaped by the fact that they're a monopoly provider of approvals for medicine. They're a chokepoint for the entire healthcare industry; if they move slowly or make mistakes, it affects everyone.
Like most monopolies, they suffer from several glaring weaknesses:
Doesn't Scale Well: A single institution making decisions that control tens of thousands of providers will miss important information and make mistakes as it struggles with the 'knowledge problem'.
No Error Correction Mechanisms: There's no one to compete with the FDA, so if they make a bad decision like approving a faulty test, or slowing down approvals for other tests, there's no alternative pathway and no competition to incentivize correction.
Can be captured: A single institution controlling a half a trillion dollar market is ripe for corruption. Critics on both the right and left allege that the FDA is in bed with pharmaceutical companies. This can look like approving drugs they shouldn't, preventing new startups from entering the market, and structuring the approval process to benefit large companies.
Over time, these forces hollow out and destroy monopoly organizations, leaving them to run zombie processes that are not effective. If it's a company, it will go out of business. If it's the single medical regulatory body in the country, it will result in deaths.
The lesson from the failures of the FDA is not that we shouldn't have certifying organizations or good controls on medical practices, it's that we need more than one. Different groups with different leadership and different incentives such that we're never again in a situation where a single complacent group can hold the fate of the country in their hands.
Conspiracy
If the FDA was such a bottleneck on responding to the pandemic, we're faced with an interesting mystery. In a world of over 7.5 billion people, living in 195 countries, why is there is a near uniformity of approach in medical regulation? While there are slightly different approaches, and some seem more competent than others, the general structure and incentives remain the same.
Defenders of the FDA will say this is evidence of its superiority. Despite the FDA's drawbacks and fuckups its the best around, imitated around the world.
But, the unanimity is suspicious. It's like hearing from someone who ended up in the suburbs of Stepford, and is looking at all the identical smiling faces and saying 'gosh this place must be perfect, look how everyone has agreed to live the same way, no horrible secret underneath that's for sure'. There are a tremendous number of free parameters in a medical approval system like length of trials, number of required participants or approvers, etc. - it's not difficult to imagine radically different systems outside of a single government bureaucracy (more on that later). It's too much of a coincidence to believe that all of these different actors randomly arrived at the same point on their own.
So, what does the convergence indicate?
A few hypotheses:
Convergent Evolution: The similar systems have evolved in this way because it's an efficient way to maximize health, safety, and public trust. Different governments arrived at this solution because their organizational structure, available technology, and trained staff were well suited for this 'globally optimum solution'. And there are advantages to global harmonization of a regulatory regime that would push companies and governments to align on the single system.
External Pressure: The similarity of the medical systems is because the FDA, and other western governments, implicity and explicitly force other governments to follow their system. As one of the largest global markets, if a foreign drug manufacturer wanted to sell their product in the US, they would need to follow US standards, enforceable by the FDA. This puts pressure on foregien governments to emulate the US.
Cultural Mimicry: Medical regulators don't exist in a vacuum, where they from first principles design a drug approval process. They exist in a world with peer companies and professional networks sharing information and opinions on 'best practices'. Remember, the incentives for any regulator in the set up are 'don't approve dangerous medicine' and 'be well-regarded'. There are systems that already exist that you can look at and copy. In this case, the FDA has had an enormous influence on foreign regulators because it was such a prominent, imitatable org.
I suspect all three are at play, but that in general we overvalue the convergent evolution argument and undervalue the influence of imitation. Regulators in different countries are copying one another - which is not a slight against the regulators, this is a human thing! Almost all companies have the same structure because if you're making a new company you've already got a hundred ways your company can fail - do you want to add tried new wacky org structure to the risks? Even if it's a good idea, you're probably better off copying the structure of organizations that exist which you can be sure work 'well enough'.
A Better Way
The key improvement is to remove the bottleneck from the system. Policy reform should be targeted at decreasing the singular, discretionary veto power of the FDA and increasing alternative approval paths for new drugs.
Repeal the Kefauver-Harris Amendment
To start we can return to a period in time when the FDA worked.
The FDA went sideways when they were tasked with evaluating and approving drugs for efficacy instead of just safety. This ballooned costs, slowed drug approvals, and greatly expanded its control over the pharmaceutical industry in distortionary ways. We can trace the testing and vaccine delays back to this 1962 law that transformed the FDA into the central planning committee of medicine.
Removing their legal mandate to test for effectiveness, and returning more of that power to other organizations - like groups of doctors, universities, and companies, would increase avenues for drug approvals. There would still be strong incentives for pharmaceutical companies to complete Phase III and Phase IV clinical trials, to provide evidence to doctors and insurance companies that the drugs worked and thus to get them prescribed and reimbursed.
The costs of this change seem minor, as the effectiveness testing by the FDA is not all that effective!
To understand how little the FDA’s two-hurdle approach contributes, consider Merck’s Keytruda (pembrolizumab), which has recently become one of the hottest drugs on the market. Keytruda got some good press after Jimmy Carter, who was suffering from melanoma, was diagnosed as cancer-free after taking it. Mr. Carter was lucky because in one clinical trial Keytruda destroyed or reduced the tumors in only 34% of patients. Keytruda—which was the fourth biggest-selling drug globally in 2018 and brought in worldwide revenues of $11.1 billion last year—is far from a sure thing.
Does the FDA, which approved Keytruda as safe and effective for melanoma, know which patients will benefit from it? No. The potential benefits described on the drug’s label are what happened to strangers in another time and place. Patients still have to try Keytruda to know if it will help them. An FDA approval for efficacy is largely duplicative.
Good Housekeeping Seal of Approval
Given, as the FDA likes to mention, it's the gold standard for drug approvals, then having an FDA seal certifying that a drug is effective at the FDA's level would be a major selling point for pharma. The FDA would not legally be able to deny access to the US market except for safety reasons, but would instead certify certain drugs as having proven their effectiveness. It's likely that insurance companies and doctors would require this seal before reimbursing or prescribing a drug, but this leaves the door open for informed individuals to use medicine that they might not otherwise have access to.
Peer regulatory approval
When the UK approved the J&J vaccine and the US didn't, why did I as an American need to solely rely on the wisdom of the US FDA? For the same reason that I can buy a Japanese car, it's reasonable to buy and use medicine/drugs that could be produced under different standards in other parts of the world.
The peer regulatory approach recognizes a number of other approving agencies as being up to a base level of competency that if an American wanted to use a drug approved by one of them, they could. A 2015 bill introduced in Congress - the RESULT Act - would have allowed for reciprocal approval of drugs, devices and biologics from certain countries including EU member countries, Israel, Canada, and Japan. It did not move forward, but the approach is a good one. A critique of the peer approval approach is that there are not many efficacious drugs in the world not approved by the FDA and giving congress a say in the vaccine approval process would politicize the approval process. While true, I believe this misses that it would improve the medical drug and device ecosystem. A startup could develop new drugs and release in multiple areas, weakening the singular veto power of the FDA.
Increased progressive approval
In a progressive approval regime drugs would be approved after they pass Phase I and Phase II testing for safety, but would continue through later Phase III and Phase IV testing, and market surveillance for side effects and efficacy. This is a slightly weaker version of the pre-1962 proposal - in this case the FDA still has authority over what drugs are in the market, but changes their evaluation methods and biases them towards action and approval, counteracting their natural bureaucratic incentives.
To a certain extent this is already happening - doctors and patients experiment with different pharmaceutical regimes to see the effects of the drug, and provide guidance to other doctors and back to the FDA. But formalizing it and speeding up access is still valuable.
Conclusion
The FDA was a major reason that the US did so poorly in fighting COVID-19, and it's continuing to undermine medical progress. We rarely see the costs that are imposed by the FDA because they are second and third order effects, bottlenecks and slowdowns as opposed to obvious blockers. If we're going to succeed against the next pandemic, or just alleviate suffering from chronic diseases, we need a better system of medical drug approvals than the one we have now.
Further Reading:
As mentioned, I think Daniel Carpenter's book on the FDA is the definitive academic history.
There's a lot to recommend from Alex Tabarrok on the FDA, consider starting with any of these.
And thank you to Sasha Chapin, Kelley Kidd, Chad Jens, Kevin Munger, Nathan Rogers, Rick Korzekwa, and everyone else who provided assistance and feedback on early drafts.